Развёртывание нейросети на базе ОС «Альт» для обнаружения онкологических заболеваний (Игорь Воронин, OSEDUCONF-2022)
Материал из 0x1.tv
- Докладчик
- Игорь Воронин


В статье обсуждаются способы развёртывания и обучения глубокой сверточной нейросети в контейнере Docker, на базе ОС Альт. Рассмотрены необходимые ресурсы для создания разных моделей распознавания результатов анализов пациентов по спектрограммам.
Предлагается решение — для определения онкологических заболеваний по спектрограммам из карт опухолевой области мозга.
Содержание
Видео
Презентация








Thesis
В современной медицине активно развиваются новые решения в области обработки и анализа данных полученных при помощи рамановской спектроскопии или спектроскопии комбинационного рассеяния — когда спектроскопический метод исследования используется для определения колебательных мод молекул и вибрационных мод в твёрдых телах. В данной работе проводится анализ спектрограмм, на основе которых можно диагностировать и различить больную ткань живого человека от здоровой. Для такой диагностики и распознавания спектров тканей была использована глубокая свёрточная нейросеть из пакета Keras — официального бэкэнда Tensorflow.
Оболочка Jupyter, делает использование Python намного проще и интуитивно понятнее даже для человека, далёкого от программирования. Существуют платные серверы, где можно развернуть и использовать данную среду, с автообновлением и регулярными backup-ами. В данной работе мы развернули нашу собственную нейросеть на серверном узле, с пропускной способностью сети гигабит в секунду: [1]
В нашем случае мы использовали уже предустановленный Doсker — сконфигурированный для развёртывания на множестве серверов.
$ docker-compose up -d
Определить адрес токена для доступа к серверу с запущенной нейросетью можно по команде:
$ docker logs tf_test
От медицинских работников были получены спектрограммы здоровых и больных тканей человека. Для обучения сети была обработана выборка порядка 1000 спектрограмм. Сеть развёртывалась в операционной системе — на российской платформе Alt p10. Основные вычисления производились на CPU сервере. Обязательным условием в нём должна быть инструкция AVX,наличие которой можно диагностировать следующей командой:
$cat /proc/cpuinfo |grep avx
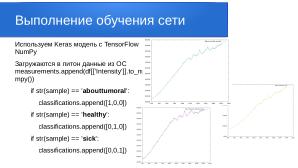
Каждый файл исходных данных содержит информацию о длине волны и интенсивности. Для разбора итоговых данных мы закодировали результаты в матрицу:
- [1,0,0] — abouttumoral (околоопухолевая область)
- [0,1,0] — healthy (здоровая область)
- [0,0,1] — sick (опухолевая область)
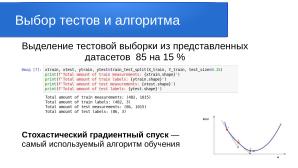
Делим датасет на тренировочную часть и тестовую в соотношении 85 к 15 параметром test_size=0.15
Были использованы предопределённые классы для слоёв:
- Dense() — полносвязный слой;
- Conv1D, Conv2D — свёрточные слои;
- MaxPooling2D, Dropout, BatchNormalization — вспомогательные слои
А также предопределённые классы моделей:
- Model — общий класс модели;
- Sequential — последовательная модель.
У каждого слоя и у модели в целом имеется свойство weights, содержащее список настраиваемых параметров (весовых
коэффициентов). В нашем случае сеть в себя включает 16,757,443 параметров.
Создаём архитектуру модели, которая является основой для определения MNIST dataset:
model2 = Sequential()
model2.add(Conv1D(128, 4, activation='relu', input_shape=(1015,1),kernel_regularizer=
regularizers.l1_l2(l1=1e-5,l2=1e-4))) # 32 neurons
model2.add(Conv1D(128, 4, activation='relu', bias_regularizer=regularizers.l2(1e-4)))
# 32 neurons
model2.add(BatchNormalization())
model2.add(Activation('relu'))
model2.add(MaxPooling1D())
model2.add(Dropout(0.25))
model2.add(Conv1D(256, 2, activation='relu', kernel_regularizer=regularizers.l1_l2(
l1=1e-5, l2=1e-4))) # 64 neurons
model2.add(Conv1D(256, 2, activation='relu', bias_regularizer=regularizers.l2(1e-4)))
# 64 neurons
model2.add(BatchNormalization())
model2.add(Activation('relu'))
model2.add(MaxPooling1D())
model2.add(Dropout(0.25))
model2.add(Flatten())
model2.add(Dense(256, activation = 'relu', use_bias=False))
model2.add(BatchNormalization())
model2.add(Activation('relu'))
model2.add(Dense(128, activation = 'relu', use_bias=False))
model2.add(BatchNormalization())
model2.add(Activation('relu'))
model2.add(Dense(64, activation = 'relu', use_bias=False))
model2.add(BatchNormalization())
model2.add(Activation('relu'))
model2.add(Dropout(0.25))
model2.add(Dense(3, activation = 'softmax'))
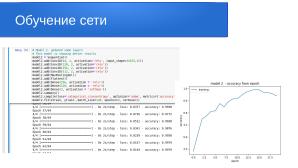
model2.summary()
model2.compile(loss='categorical_crossentropy',optimizer='adam',metrics=['accuracy'])
model2_hist = model2.fit(xtrain, ytrain ,batch_size=128, epochs=100, verbose=1)
Total params: 16,757,443
Trainable params: 16,755,779
Non-trainable params: 1,664
Epoch 1/100 1/1 [=============================] - 2s 2s/step - loss: 1.7923 - accuracy: 0.2321 Epoch 100/100 1/1 [=============================] - 1s 713ms/step - loss: 0.3576 - accuracy: 0.9107
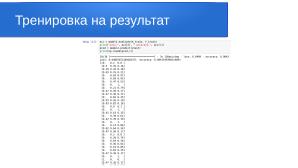
Чтобы оценить итоговую точность модели на тестовой части датасета, выполняем следующие команды:
acc = model2.evaluate(xtest, ytest)
print("Loss:", acc[0], "Accuracy:", acc[1])
pred = model2.predict(xtest)
print(np.round(pred,2))
1/1 [=============================] - 0s 229ms/step - loss: 9.8597 - accuracy: 0.9636
[[0.09 0.91 0. ]
[0.01 0.99 0. ]
[0. 1. 0. ]
[0. 1. 0. ]
[0.1 0.9 0. ]]

Полученный результат говорит о том, что тестовые спектрограммы были распознаны с вероятностью 91 % для здоровых тканей.
!.jpg)